・Genomic Analysis for Precision Medicine in Coronary Artery Disease
Background
Coronary artery disease is a generic term for diseases such as myocardial infarction and angina pectoris that cause narrowing or blockage of the coronary arteries that supply nutrition to the heart, and behind these diseases exist conditions such as hypertension and dyslipidemia that progress atherosclerosis. As coronary artery disease is the world’s leading cause of death, there is an urgent need to understand the causes of the disease and establish effective prevention and treatment methods.
Coronary artery disease is highly heritable [1] and many studies have been conducted and many associations between genetic variants and coronary artery disease have been reported. For PCSK9 gene, which has been shown to be strongly associated with coronary artery disease, we have successfully developed therapeutic drugs that target it. In addition, it has recently become clear that the Genetic Risk Score (GRS) [2], which is based solely on genetic information, can predict disease onset with high accuracy.
Thus, understanding the origins of the disease, developing therapeutics and predicting the risk of developing the disease based on genetic information is expected to play an important role in advancing medical science and medicine for coronary artery disease in the future. However, previous studies have focused primarily on European populations, and it is not clear whether the results of these studies are applicable to East Asian populations, including the Japanese. It is also known that there are large ethnic differences in the distribution of genetic variation, but the association between genetic variation that does not exist in European populations and coronary artery disease has not been well studied.
Besides, the association between genetic variants and coronary artery disease, which does not exist in European populations, has not been well studied. To this end, the international research group searched for genetic variations in the development of coronary artery disease in approximately 170,000 subjects enrolled in the Biobank Japan.
Methods and Results
Comparing the genome sequences of approximately 25,892 coronary artery disease patients in the Biobank Japan and 142,336 controls, a total of approximately 170,000 people, the international collaborative research group conducted a genome-wide association analysis (GWAS) [3] to comprehensively detect the genetic variations associated with coronary artery disease. This study is the largest GWAS project on coronary artery disease in a non-European population in the world.
Prior to conducting the GWAS, we first developed a new reference panel [4] for genotype imputation [5] using whole genome sequencing [6] data from 4,417 Japanese individuals. This reference panel contains a large number of rare variants specific to the Japanese [7], and allowed us to infer about twice as many genetic variants as before.
Using this densely estimated genetic information, GWAS identified 48 coronary artery disease susceptibility loci and 73 genetic variants across the genome; eight of the 48 disease susceptibility loci had not been identified in previous studies of European populations. A missense mutation in the RNF213 gene on chromosome 17 [8] was previously known to be a causal mutation in the gene responsible for moyamoya disease [9] seen in children. Previous studies in Japan have reported an association with coronary artery disease, but this study was the first to show an association with coronary artery disease within the framework of GWAS. The reason for this is that this missense mutation was not present in the European population and could not be evaluated in previous GWASs on European populations.
Typically, the odds ratio of disease-associated genetic variants found in GWAS is mostly small, ranging from 1.1-1.2. This is due to the fact that previous GWASs have not been able to accurately estimate the pathogenicity of genetic variants with a population frequency of less than 1%. In the present study, we used a reference panel that is specific to the Japanese population, and thus were able to accurately estimate the pathogenicity of low to very low frequency variants (<1% population frequency). The strongest pathogenicity was observed in a genetic mutation on LDLR gene (stop-gain mutation [8], LDLR p.K811X, 0.038% population frequency) that substantially reduces the function of LDL receptors, which are important for cholesterol metabolism. This mutation had a strong effect on the risk of developing coronary artery disease (odds ratio [10]) of 5 (Figure 1). Carrying this mutation could be interpreted as being five times more likely to develop coronary artery disease than those who did not carry it. We also found that carrying this mutation increased serum total cholesterol levels by an average of 57 mg/dL.
On the other hand, mutations that are protective against coronary artery disease were also detected: the PCSK9 gene is known to promote the development of coronary artery disease by interfering with the function of LDL receptors. Carrying a genetic mutation that reduces the function of the PCSK9 gene (missense mutation, PCSK9 p.R93C) decreased an odds ratio of 0.6, and cholesterol levels by an average of 20 mg/dL (Figure 1). Furthermore, we found that these mutations were unique to the Japanese and not found in other ethnic groups.
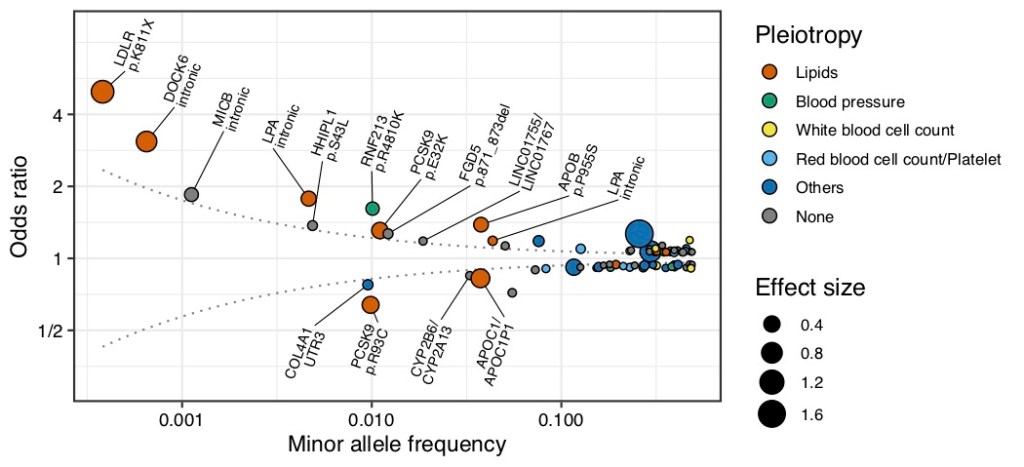
Next, the results of the Japanese GWAS (approximately 170,000) were combined with those of the European population (approximately 180,000 from the CARDIoGRAMplusC4D study [11] and 300,000 from the UK Biobank [12]) to create one of the world’s largest trans-ethnic GWAS in coronary artery disease with a total of more than 600,000 people. We have identified 175 disease susceptibility loci that are associated with coronary artery disease. Thirty-five of these regions were newly identified, including the HMGCR gene, which is a target of the most important therapeutic drug in coronary artery disease. Thus, the susceptibility loci found in this study may contain genes that are useful as therapeutic drug targets for coronary artery disease, and will provide important information for future drug development.
We then used the results of the GWAS obtained in this study to create a GRS and examine its predictive performance in the Japanese genomic cohort (JPHC study, J-MICC study and OACIS study). It has long been known that GRS calculated from GWAS results for a specific ancestry population do not fit other populations and have poor predictive performance. In this study, it was confirmed that the GRS calculated from the GWAS results for European populations had lower predictive performance than the GRS calculated from the GWAS results for Japanese. When the trans-ethnic GWAS results were used to create the GRS, the performance of the trans-ethnic GRS was found to be superior to the Japanese GRS and the European GRS (Figure 2).
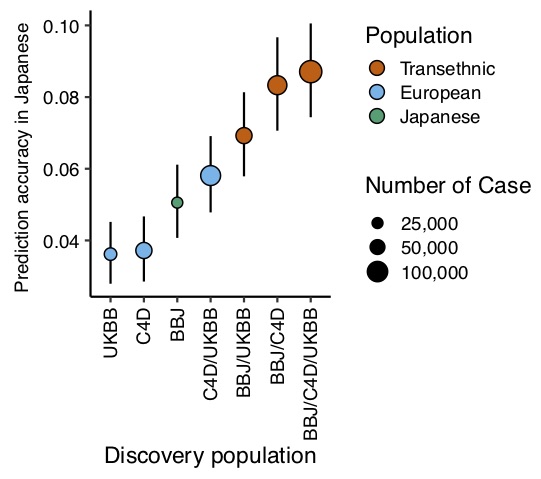
Since GWAS has been conducted mostly for European populations, the accumulation of data for European populations is large, but the accumulation of data for non-European populations, including Asians, is small, and the benefit of risk prediction by the GRS for non-European populations has been limited. Our results showed that the integration of existing data will be useful in the future development of the GRS in non-European populations.
Future
The newly identified disease susceptibility loci identified in this study will provide important information for future understanding of the pathology of coronary artery disease and the discovery of therapeutic agents. In addition, GRSs optimized for Japanese people are expected to be used effectively in the future of precision medicine based on genetic information. The results of this study are freely available for research purposes via the National Bioscience Database Center [13] (https://humandbs.biosciencedbc.jp/hum0014-v20)
Key words
[1] Hereditary
A disease is said to be “highly heritable” when it is more common within a blood relative. Diabetes and coronary artery disease are known to be relatively highly heritable general complex diseases.
[2] Genetic Risk Score (GRS)
An estimate of an individual’s susceptibility to a disease calculated by adding up the effects of tens of thousands to millions of genetic variations in the genome. In the case of coronary artery disease, the higher this value, the greater the likelihood of developing the disease. GRS is an abbreviation of the Genetic Risk Score.
[3] Genome-wide association analysis (GWAS)
A comprehensive method of detecting genetic mutations that affect the development of a disease. GWAS was first reported by RIKEN in 2002. GWAS is an abbreviation of the Genome Wide Association Study.
[4] Reference Panel
Whole-genome sequencing data to be used for genotype imputation. In the past, data generated by the 1000 Genome Project and other projects were often used, but with the generalization of whole-genome sequencing, ancestry- and disease-specific reference panels are becoming available.
[5] Genotype imputation
A method of using the results of whole genome sequencing to estimate genetic variation that is not detected by the commonly used microarray genetic variation analysis. It can increase the number of mutations that can be used for genetic analysis.
[6] Whole Genome Sequencing
A comprehensive method of detecting genetic variation on a genome. Previously, sequencing the entire genome of a single person required a budget on the scale of a national project, but advances in technology have made it possible to do this on a large scale.
[7] Rare variants
A genetic mutation that has a frequency of less than 1% in a population. Because mutations with strong effects are less likely to be spread within a population, mutations with high pathological significance tend to be less frequent.
[8] Missense and stop-gain mutations
A mutation in the genomic region that stops the translation into proteins is called a “stop-gain mutation”. This mutation produces a shorter protein than the original one, which has a very large impact on its function. A “missense mutation” causes amino acid substitution in the protein during translation, but only a partial substitution, so the effect varies depending on the protein site and the nature of the substitution.
[9] Moyamoya disease
It is a disease first discovered in Japan in which collateral vessels develops due to progressive occlusion of the Willis arterial ring in the cerebral blood vessels. It is a common cause of stroke in children.
[10] Odds ratio
A measure of the magnitude of the risk of developing the disease. It expresses the number of times the risk of developing the disease increases relative to a reference.
[11] The CARDIoGRAMplusC4D study
This is the world’s largest genomic study of coronary artery disease. It is led by U.S. researchers, who are integrating and analyzing a large number of genomic cohorts. Many of the discoveries in genomic study in coronary artery disease are rooted in this work. CARDIoGRAMplusC4D is the abbreviation of the Coronary ARtery DIsease Genome wide Replication and Meta-analysis (CARDIoGRAM) plus The Coronary Artery Disease (C4D) Genetics.
[12] UK Biobank
It is a genomic biobank established and maintained in the United Kingdom that collects genomic and medical information on participants. If a researcher applies for it, he or she is free to conduct genomic research within the limits of personal information protection.
[13] National Bioscience Database Center (NBDC)
It is a national database to collect, share, organize, and integrate big data generated from domestic life science research. Research results from Biobank Japan are available through NBDC. For more information, please visit NBDC’s website (https://biosciencedbc.jp/)
